


 SCM
SCM
SCM home > MATERIAL Sci > SCM
home > MATERIAL Sci > SCM

분자동역학, 몬테카를로 시뮬레이션, Ab initio


- Amsterdam Modeling Suite
-
Powerful Computational Chemistry
Making Computational Chemistry Work for You
자세한 문의는 02-838-4016 또는 comj@tnjtech.co.kr로 요청 주시기 바랍니다.

 Powerful molecular DFT to understand chemistry
Powerful molecular DFT to understand chemistry최적화된 분자 구조, 전이상태 구조, 반응성, 다양한 스펙트럼을 DFT를 이용하여 예측할 수 있습니다.
상대성 효과를 고려하면서 주기율표의 모든 원소들을 지원하고 수준 높은 정확성으로 계산할 수 있는 STO all-electron basis sets을
제공하여 중원자들이 포함된 전이금속착물, 나노입자, 유기적 전자공학 분야 연구에 많이 활용됩니다.

주요 기능
- Modern xc functionals, including latest dispersion corrections, double hybrids, and range-separated hybrids
- Self-consistent spin-orbit coupling TDDFT
- Charge transfer integrals, NEGF
- Many bonding analysis tools (EDA, ETS-NOCV, QTAIM, NCI)
- Fast G0W0 and RPA single point calculations
- QM/MM and QM/QM' calculations of arbitrary periodicity
- Slater-type orbitals: correct nuclear cusp (NMR, EPR)
- Environments and solvation: DIM/QM, FDE, COSMO, SM12
- Self-consistent spin-orbit coupling TDDFT
- Charge transfer integrals, NEGF
- Many bonding analysis tools (EDA, ETS-NOCV, QTAIM, NCI)
- Fast G0W0 and RPA single point calculations
- QM/MM and QM/QM' calculations of arbitrary periodicity
- Slater-type orbitals: correct nuclear cusp (NMR, EPR)
- Environments and solvation: DIM/QM, FDE, COSMO, SM12
 Periodic DFT for nanotubes, surfaces, and bulk
Periodic DFT for nanotubes, surfaces, and bulkAccurate periodic density functional theory code로 periodic DFT 계산에 atomic orbitals을 사용하여 plane waves 방식보다 proper treatment of surfaces, efficient computations of sparse matter 등 직접적이고 세밀한 분석 방법을 제공합니다. 계산 속도 면에서 빠른 계산이 필요할 경우를 위해 plane wave code인 Quantum ESPRESSO를 함께 제공합니다.
주요 기능
- Model surfaces & nanowires as true 2D & 1D periodic systems
- All-electron basis sets for all elements
- Easy to treat core holes (X-ray)
- Relativistic effects treated efficiently and accurately with scalar ZORA
- Self-consistent spin-orbit coupling
- Homogeneous electric fields
- Continuum solvation with COSMO & SM12
- Calculate many spectra, orbitals & density properties
- DFT-1/2 and model potentials (KTB-mBJ, GLLB-sc) for accurate band gaps
- All-electron basis sets for all elements
- Easy to treat core holes (X-ray)
- Relativistic effects treated efficiently and accurately with scalar ZORA
- Self-consistent spin-orbit coupling
- Homogeneous electric fields
- Continuum solvation with COSMO & SM12
- Calculate many spectra, orbitals & density properties
- DFT-1/2 and model potentials (KTB-mBJ, GLLB-sc) for accurate band gaps
 Fluid thermodynamics from quantum mechanics
Fluid thermodynamics from quantum mechanicsCOSMO-RS (COnductor-like Screening MOdel for Realistic Solvents) 모듈은 양자역학 데이터를 기반으로 유체와 용액의 열역학적
물성들을 예측합니다.
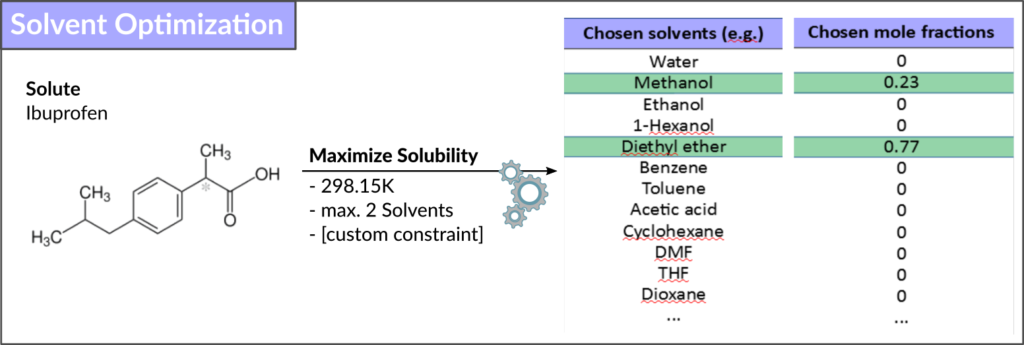
주요 기능
- Solubilities, partition coefficients (log P, log kOW)
- pKa values (empirical fit improves predictions)
- Activity coefficients, solvation free energies, Henry's law constants
- Vapor pressures, boiling points, vapor-liquid diagrams binary and ternary mixtures (VLE/ LLE)
- Excess energies, azeotropes, miscibility gaps
- Composition lines, flash points
- Optimizing solvent mixtures for solubility or extraction
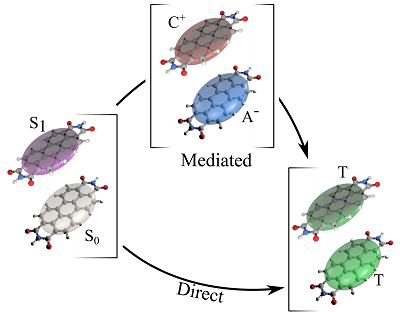
 Fast approximate DFT for molecules, 1D, 2D and 3D
Fast approximate DFT for molecules, 1D, 2D and 3DDensity-Functional based Tight-Binding (DFTB)은 미리 계산된 파라미터들과 minimal basis, nearest-neighbor interaction만을 고려하여 DFT 계산보다 훨씬 적은 계산 시간으로 큰 시스템을 DFT 결과에 준하는 비교적 정확하게 계산합니다.
주요 기능
- Optimize minima and transition states and quickly characterize them
- Periodic optimization under pressure
- Fast UV/VIS spectra and excited state geometries
- Doubly parallelized IR and VCD spectra, phonons, stress/strain
- Band structures, effective mass, Density of States, pDOS, Molecular Orbitals
- Advanced Molecular Dynamics and Monte Carlo
- Solvation with GBSA
- Charge transport with NEGF
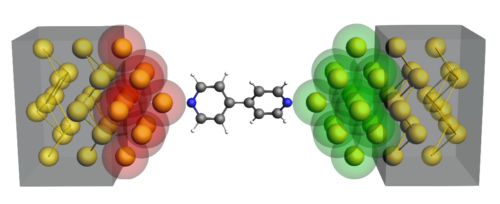
 Reactive MD with GUI and analysis tools
Reactive MD with GUI and analysis toolsReactive force field를 이용하여 화학 반응이 일어나는 large-scale systems을 연구할 수 있습니다.
Reactive force field의 개발자인 van Duin 교수 연구그룹과 공동 개발하여 original ReaxFF code를 병렬화 및 최적화하여 수십만개의 원자로 구성된 혼합물 시스템도 일반 데스크톱 컴퓨터에서 계산할 수 있습니다.
Classical force field에서 다루기 어려웠던 전이금속들도 다룰 수 있고 다양한 조합으로 80개 이상의 ReaxFF force field를 제공합니다.
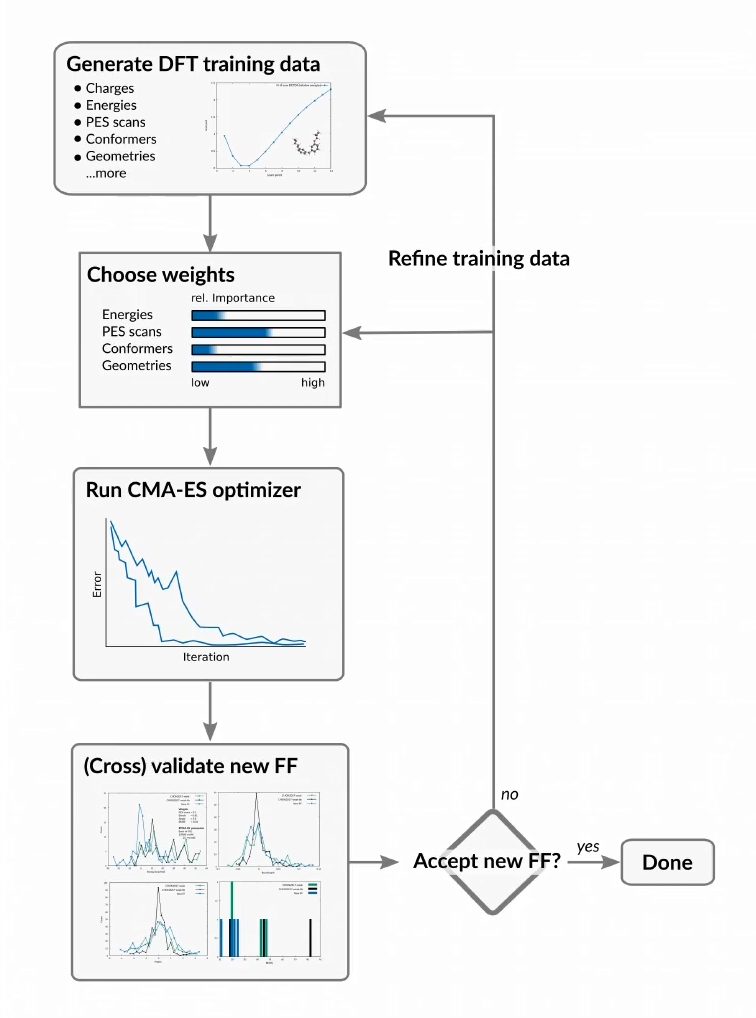
특화된 모델 시스템에 맞춰 새로운 ReaxFF parameter sets을 개발하거나 기존의 parameter set을 개선할 수 있도록 (re)parametrization tool을 제공합니다.
- Mechanical properties of epoxide polymer
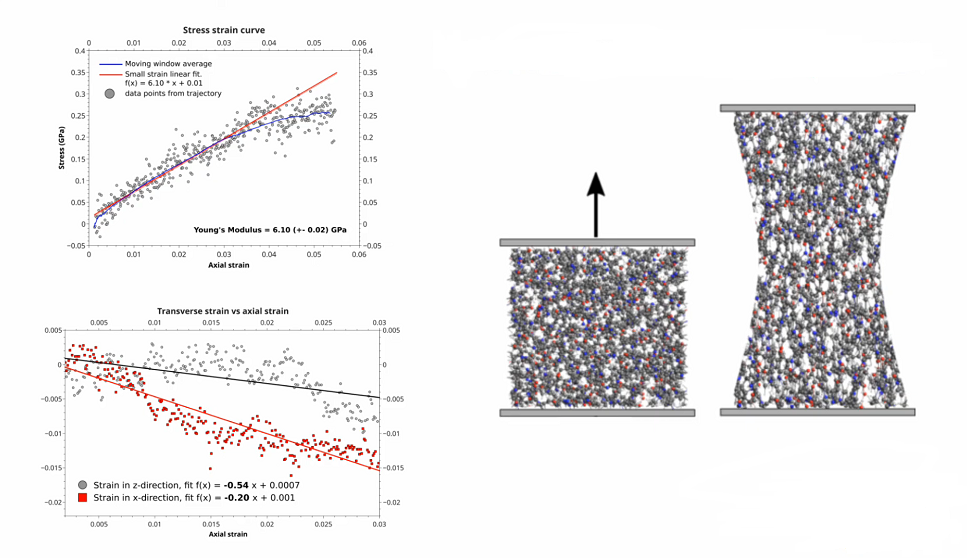
- Fitting ReaxFF force field parameters with CMA-ES
주요 기능
- Analyze changing molecular composition and reaction pathways (ChemTraYzer), track surface reactions
- AMStrain: Create and manage training sets for ReaxFF re-parametrization
- Accelerate bond breaking with Collective Variable-driven Hyperdynamics
- Molecule gun and molecule sink: depositing atoms and molecules, CVD, sputtering processes
- Bond boost for cross-linking polymers
- Grand-Canonical Monte Carlo: reactivity under thermodynamic equilibrium conditions
- Force-bias Monte Carlo
- Various free energy methods and MD analysis via the PLUMED library
- Hybrid parallelization (MPI, openMPI)
- Thermal conductivity (T-NEMD)
- ACKS2 charge equilibration: correct long-range charge behavior (batteries, enzymes)
- eReaxFF: explicit electrons
 Machine Learning Potential
Machine Learning Potential힘장을 사용하는 시뮬레이션 연구에 필요한 계산 시간 정도의 비용으로 분자동역학 시뮬레이션, 포텐셜 에너지 표면을 탐색할 수
있는 machine learning potential 입니다.
사전 매개 변수화 된 모델 ANI-1ccx, ANI-2x를 제공하여 바로 계산에 활용할 수 있고 PiNN, SchNetPack, sGDML, TorchANI 엔진을
이용하여 새로운 포텐셜을 만들 수 있습니다.

 |